Kategorien durchsuchen
Entdecken
Fiverr Pro
Deutsch
$
USD



Automatische Übersetzung
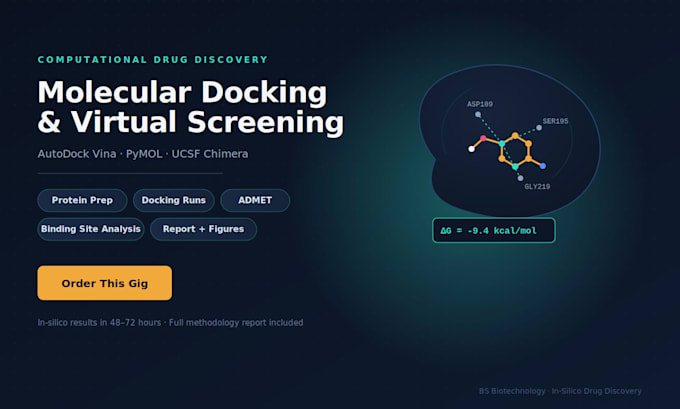
Ich bin ein Forscher im Bereich der computergestützten Biologie mit praktischer Erfahrung in strukturorientierter Wirkstoffentdeckung. Meine Abschlussarbeit beschäftigte sich mit in silico Inhibitoren für die CARDS-Toxin von Mycoplasma pneumoniae, bei der ich aus großen Wirkstoffbibliotheken mit AutoDock Vina zwei Leitverbindungen identifizierte, die durch ADMET- und Lipinski/Veber-Pharmakokinetik-Filter validiert wurden.
Was ich anbiete:
Zielproteinvorbereitung aus PDB
Molekulardocking mit AutoDock Vina
Virtuelle Screening von Wirkstoffbibliotheken
ADMET- & Drug-Likeness-Bewertung (Lipinski, Veber)
Ligand-Protein-Interaktionsanalyse (PyMOL)
Klare, publikationsreife Berichte mit Visualisierungen
Tools: AutoDock Vina, PyMOL, UCSF Chimera, RCSB PDB, ChEMBL, SwissADME
Schreib mir vor der Bestellung, um dein Zielprotein und den Projektumfang zu besprechen.
molecular docking, Ai Driven Drug Discovery,3D models
Sprachen
Automatische Übersetzung