Kategorien durchsuchen
Entdecken
Fiverr Pro
Deutsch
$
USD
Leidenschaftlich beim Lernen
Über diesen Service
Arbeitest du an einem Protein-Ligand-System und brauchst zuverlässige, gut analysierte molekulardynamische Simulationen für deine Forschung oder Veröffentlichung? Ich biete End-to-End-AMBER-MD-Simulationsdienste vom Systemaufbau bis zu publikationsfertigen Abbildungen, basierend auf derselben Pipeline, die in peer-reviewed Forschungsarbeiten zur computergestützten Wirkstoffentdeckung verwendet wird.
Ich bin ein Forscher im Bereich der computergestützten Biologie mit praktischer Erfahrung in AMBER/AmberTools, cpptraj und Python-basierten Analyse-Workflows. Meine Arbeit hat direkt zu Manuskripten beigetragen, die in peer-reviewed Fachzeitschriften in der computergestützten Pharmakologie eingereicht wurden.
Was ich liefere
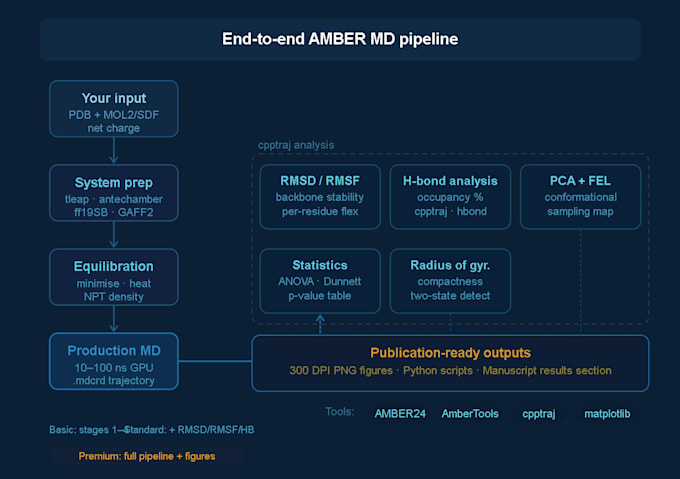
Systemeinrichtung
Vorbereitung der Proteinstruktur und Protonierungszustand
Liganden-Parameterisierung mit antechamber
Solvation (TIP3P- oder OPC-Wasserbox), Zugabe von Gegenionen
Kraftfeldzuweisung: ff19SB (Protein), GAFF2 (Kleinstmolekül)
Minimierung, Erhitzung und NPT/NVT-Äquilibrierung
Produktions-MD-Lauf
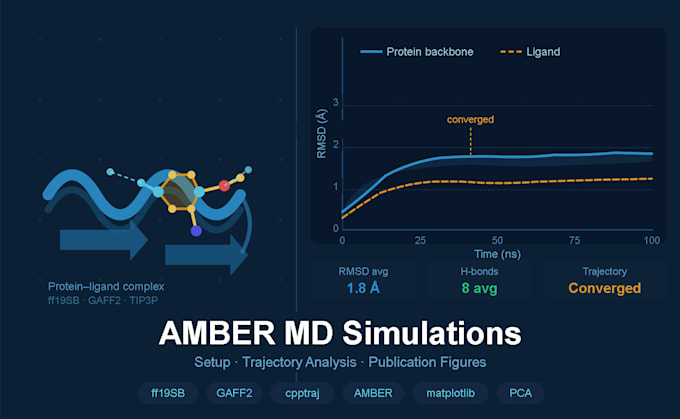
10, 50 oder 100 ns Produktionstrajektorien (abhängig vom Paket)
Vollständige .mdcrd-Trajektor-Dateien geliefert
Trajektorienanalyse (Standard & Premium)
Vollständige, publikationsfertige Abbildungen
Warum mit mir arbeiten?
Reale amber-Erfahrung, kein Cloud-Wrapper oder Online-Tool.